زنگ تفریح 194، ساختار الکترونی، مدل موجی، پیوند والانس، اربیتال مولکولی، نظریهی اساسی چگالی، دینامیک شیمیایی، دینامیک شیمیایی آدیاباتیک (بیدررو)، دینامیک شیمیایی غیر آدیابیتیک
ساختار الکترونی
مدل موجی
پایهی مکانیک کوانتومی و شیمی کوانتوم مدل موجی میباشد که در آن اتم یک هستهی کوچک و چگال با بار مثبت است که با الکترونها احاطه شده، بر خلاف مدل بور دربارهی اتم. البته مدل موجی الکترونها را به صورت ابرهایی که در اربیتالها حرکت میکنند وصف میکند که جایگاه این ابرهای الکترونی توسط توزیع احتمال که از نقاط گسسته بهتر است بیان میشود. مزیت این مدل متکی به قدرت پیشگویی آن است؛ به خصوص این مدل، مدل شیمیایی عناصر دیگر در جدول تناوبی عناصر را پیشبینی میکند. مدل موجی به این دلیل به این نام خوانده میشود که ویژگیهای نشان داده شدهی الکترون (مانند تداخل) از قدیم با امواج در ارتباط بوده.
پیوند والانس
| اگرچه پایهی ریاضیاتی شیمی کوانتوم توسط شرودینگر در سال 1926 گزارده شده است، این مورد پذیرفته شده است که اولین محاسبهی درست در شیمی کوانتوم از فیزیکدانان آلمانی والتر هایتلر (Walter Heitler) و فریتز لوندون (Fritz London) بر روی مولکول هیدروژن (H2) در 1927 بوده است. روش هایتلر و لوندون توسط فیزیکدان نظری آمریکایی جان سی.اسلتر (John C.Slater) و شیمیدان نظری آمریکایی لینوس پائولینگ (Linus Pauling) تعمیم یافت تا تبدیل به روش پیوند والانس شود (یا روش هایتلر-اسلتر-پائولینگ (HLSP)). در این روش، توجه اصولا به سمت برهمکنشهای دوبهدوی میان اتمها است و در نتیجه این روش بسیار وابسته به طرح قدیم از پیوندها در شیمی کلاسیک میباشد. |
 |
اربیتال مولکولی
| یک پیشرفت نسبی در 1929 توسط فردریک هوند (Friedrich Hond) و رابرت اس.مولیکن (Robert S.Mulliken) ایجاد شد که الکترونها توسط توابع ریاضیاتی به صورت غیر مستقر در مولکول توصیف شدند. نظریهی هوند-مولیکن یا روش اربیتال مولکولی کمتر ذاتا شیمیایی است ولی خاصیت پیشبینی خواص طیف نگاری را بهتر از روش VB دارا است. این پیشرفت پایهی ادراکی روش Hartee-Fock و بعدا روش post Hartee-Fock شد. |
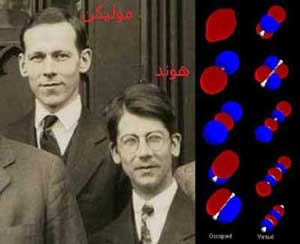 |
نظریهی اساسی چگالی
مدل Thomas-Fermi بصورت مستقل توسط توماس (Thomas) و فرمی (Fermi) در سال 1927 ارائه شد. این اولین تلاش برای توضیح سیستمهای پرالکترون بر پایهی چگالی الکترونی به جای توابع موجی بود، اگرچه در طرز عمل تمامی مولکولها چندان موفق نبود. این روش پایهی چیزی را که اکنون به عنوان نظریهی اساسی چگالی میشناسیم ایجاد کرد. هر چند این روش کمتر از روشهای post Hartee-Fock گسترش یافت، نیاز کم قابل توجه آن به محاسبات باعث میشود که از عهدهی مولکولهای چند اتمی بزرگتر و حتی ماکرو مولکولها برآید. این مدیریت محاسباتی و بعضی اوقات قابل مقایسه با MP2 و CCSD (روشهای post Hartee-Fock) آن را به یکی از مشهورترین روشهای محاسباتی در حال حاضر تبدیل کرده.
دینامیک شیمیایی
یک مرحله فراتر میتواند شامل حل معادلهی شرودینگر توسط molecular Hamiltonian برای مطالعهی حرکت مولکولها شود. حل مستقیم معادلهی شرودینگر دینامیک مولکولی کوانتومی خوانده میشود، مطابق تقریب نیمه کلاسیک دینامیک مولکولی نیمه کلاسیک و مطابق علم مکانیک کلاسیک در چارچوب دینامیک مولکولی (MD) میباشد. تقریبهای آماری مورد استفاده برای مثال در روشهای Monto Carlo، نیز ممکن میباشند.
دینامیک شیمیایی آدیاباتیک (بیدررو)
| در علم دینامیک آدیابیتیک، برهمکنشهای درون اتمی توسط یک پتانسیل اسکالر به نام سطوح انرژی پتانسیل نمایش داده میشوند. این تقریت بورن-آپنهایمر (Born-Oppenheimer) است که توسط بورن (Born) و آپنهایمر (Oppenheimer) در سال 1927 معرفی شد. کاربردهای پیشرو این تقریب در شیمی توسط رایس (Rice) و رمسپرجر (Ramsperger) در 1927 و کسل (Kassel) در 1928 ایجاد شد و به صورت نظریه RRKM در سال 1952 توسط مارکوس (Marcus) (که نظریهی حالت گذار که توسط آیرینگ (Eyring) در سال 1935 گسترش یافته بود را مد نظر قرار داده بود) عمومیت یافت. این روشها تخمینهای سادهی سرعتهای واکنش تک مولکولی از یک سری مشخصات اندک از سطح پتانسیل را ممکن ساختند. |
 |
دینامیک شیمیایی غیر آدیابیتیک
دینامیک غیر آدیابتیک شامل برهمکنشهای میان چندین سطح انرژی پتانسیل جفت شده (مطابق با حالتهای مختلف کوانتوم الکترونی از مولکول) میشود. شرایط جفتشدگی vibronic coupling نامیده میشود. این تحقیق ابتدایی در این شاخه توسط استاکلبرگ (Stuckelberg)، لاندو (Landau) و زنر (Zener) در دههی 1930 انجام شد. تحقیق آنها به عنوان گذار Landau-Zener شناخته میشود. رابطهی آنها این امکان را برای احتمال گذار میان دو منحنی پتانسیل آدیاباتیک در همسایگی یک avoided crossing برای محاسبه ایجاد میکند.